
厚生労働省は7月24日、中央社会保険医療協議会(中医協)の薬価専門部会(部会長=中村洋・慶應義塾大大学院経営管理研究科教授)を開き、2020年度の薬価制度改革に向けて、関係団体からのヒアリングを実施した。【新井裕充】
意見陳述と質疑の模様は、以下のとおり。
※ このページは意見陳述。質疑は次のページ。
質疑のページはこちら

〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
はい、ではただいまより、第154回「薬価専門部会」を開催いたします。
まずは本日の出欠状況についてご報告します。本日は、全員がご出席になります。
引き続き、厚生労働省におきまして異動がございましたので、事務局より紹介をお願いいたします。
では医療課長、森光さん、お願いいたします。
〇厚労省保険局医療課・森光敬子課長
はい。事務局より異動のご紹介をさせていただきます。
医政局・林(俊宏)経済課長でございます。
〇厚労省医政局経済課・林俊宏課長
林でございます。よろしくお願いいたします。
〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
ありがとうございました。
なお、会議冒頭のカメラの頭撮りはここまでとさせていただきます。
それでは、議事に入らせていただきます。
今回は、関係業界からの意見聴取を行いたいと思います。
関係業界として、
日本製薬団体連合会
米国研究製薬工業協会
欧州製薬団体連合会
日本医薬品卸売業連合会
再生医療イノベーションフォーラム
日本バイオテク協議会
より意見を聴取したいと考えております。
それでは早速、意見陳述に移りたいと思います。
まずは関係団体の皆さまよりプレゼンテーションしていただき、その後に質疑とフリーディスカッションを行いたいと思います。
それではまず、日本製薬団体連合会、米国研究製薬工業協会および、欧州製薬団体連合会の順に自己紹介を行った上で、プレゼンテーションをお願いいたします。
では、よろしくお願いいたします。

〇日本製薬団体連合会・多田正世会長代理(大日本住友製薬代表取締役会長)
おはようございます。私は、日本製薬団体連合会の多田と申します。意見陳述のお時間を頂き、誠にありがとうございます。
本日は、手代木功会長(塩野義製薬代表取締役社長)の代理といたしまして、業界意見を述べさせていただきます。

はじめに私から、製薬企業を取り巻く環境や次期薬価制度改革に向けての意見の概要について述べた後に、
新薬につきましては、日本製薬工業協会の中山会長(第一三共代表取締役会長)、
後発品につきましては、日本ジェネリック製薬協会の澤井会長(沢井製薬代表取締役社長)より、陳述をさせていただきます。
(スライド)次、お願いいたします。

日薬連は、業態別団体(15団体)と、地域別団体(16団体)で構成される連合会でございまして、このスライドは、業態別団体の位置付けを模式的に示しております。
次、お願いいたします。
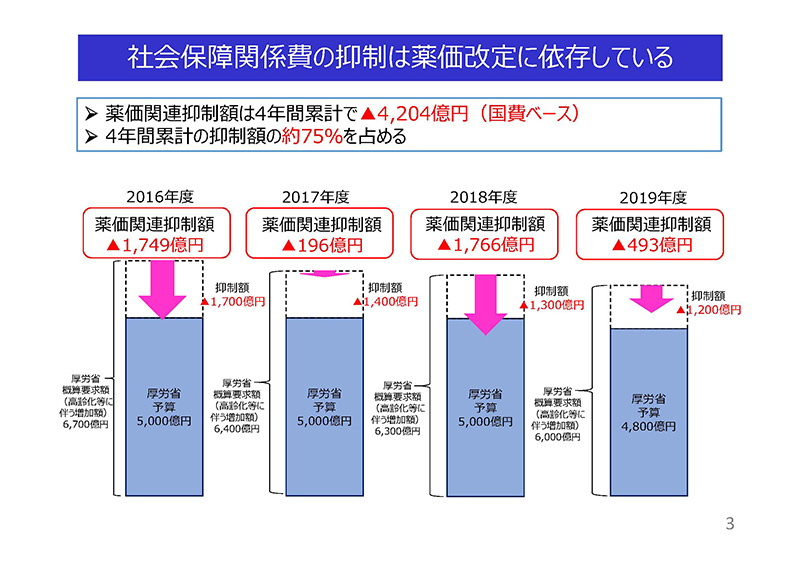
こちらは、社会保障関係費の抑制が薬価改定に依存していることを示した図でございます。
薬価関連抑制額は、予算上の国費ベースにおいて、4年間累計でマイナス4,200飛び4億円であり、抑制額の約75%を占めております。
国民皆保険制度の堅持は最重要課題と認識はしておりますけれども、薬価制度の見直しのみで財源を捻出することはもはや限界にあり、広い視点から検討すべきだと(いうことで)ございます。「限界である」というふうに感じておるわけでわけでございます。
スライド4でございますが、
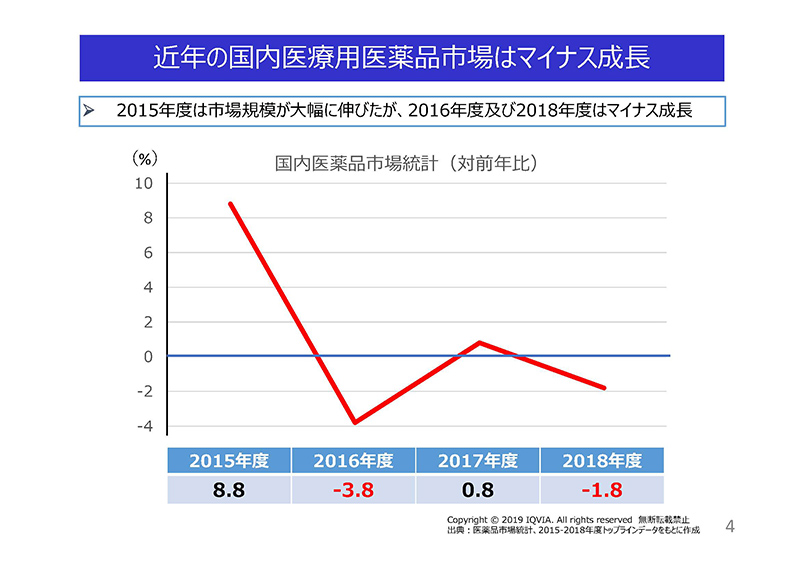
こちらは近年の国内の医療用医薬品市場の対前年度比の推移でございます。
2015年度は、画期的なC型肝炎治療薬等の上市によりまして、国内市場は大きく伸びましたが、その後は、特例拡大再算定の導入や、薬価制度の抜本改革、後発医薬品の使用促進等により、国内市場はマイナス成長となりました。
5ページをお願いいたします。
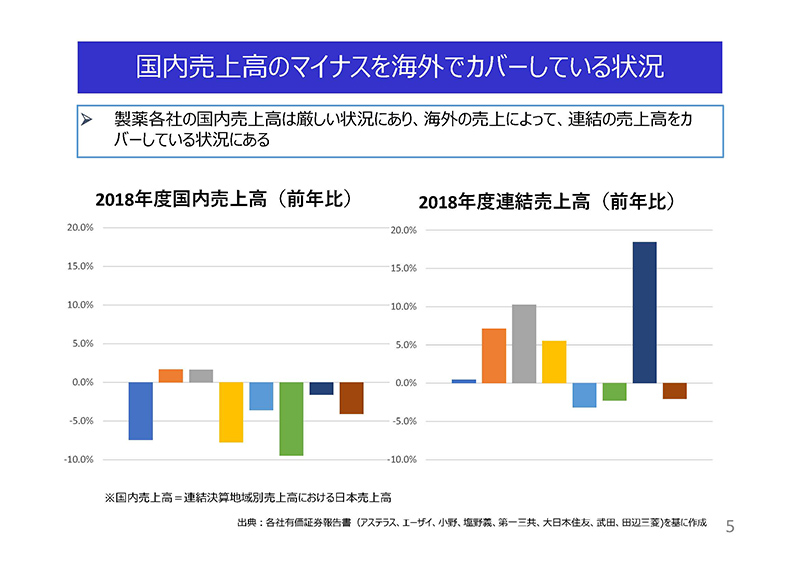
国内製薬企業8社の2018年度の、
左のグラフが度国内売上高、および右グラフは連結売上高の前年比を示しております。
製薬各社の国内売上高は厳しい状況にあり、海外の売上によって連結の売上高をカバーしている状況にございます。
6ページをお願いいたします。

こちらは、製薬会社8社における国内従業員数の状況でございます。
製薬会社単体(8社)における国内従業員数は3年連続で減少しており、各社では事業効率化のため再編に伴う分社化、工場の譲渡および閉鎖、早期希望退職者の募集などが行われている状況にございます。
スライドの7をお願いいたします。

2018年度以降の薬価制度をめぐる動向について改めて振り返りますと、
薬価制度の抜本改革では、イノベーションの推進については重要視されておらず、結果として、薬価を引き下げる方向に偏ったものになったと言わざるを得ません。
また、消費税率引上げに伴う薬価改定では、半年前、半年間前倒しで実勢価に基づく引下げが行われるために、製薬企業の経営等に対する影響は極めて大きいと考えます。
さらに費用対効果の制度化では、有用性系加算が最大90%の引下げになるなど、「薬価制度補完をする仕組み」と言うにはあまりにも厳しい内容を含むものとなりました。
こうした製薬企業を取り巻く環境が厳しくなる中、次期薬価制度改革によってイノベーションが推進され、医療の質の向上に資する制度へと改善されるよう、私どもの意見を述べさせていただきます。
スライドの8をお願いいたします。

まず新薬につきましては、
・新薬創出等加算の品目要件の拡充や企業要件の見直し、
・薬価算定における類似薬の対象を拡大する仕組み、
・薬価収載時および収載後の評価等についての検討
が必要と考えております。詳細につきましては後ほど、中山会長よりご説明させていただきます。
次に、長期収載品と後発品および基礎的医薬品等についてでございます。

長期収載品にかかるG1、G2ルールは、平成30年度改定で導入されたところであり、新薬の研究開発から承認、上市に至るまでには長期の期間が必要であること、
また、長期収載品から後発品への置換えや安定供給等に与える影響が現時点では不透明であることを踏まえまして、長期収載品の段階的引下げまでの期間について拙速に見直すべきではないと考えます。
G1、G2ルールによる薬価の引下げは、個別品目や企業に対して大きな影響を与えるため、安定供給という観点から、引下げ率の下げ止めや、影響の大きい企業への円滑実施措置について継続すべきと考えます。
後発品につきましては後ほど、澤井会長よりご説明をさせていただきます。
最後に、基礎的医薬品等でございます。
医療上必要な医薬品の継続的な安定供給を確保する観点から、基礎的医薬品については、対象範囲のさらなる拡充や要件の見直しが必要と考えます。
あわせて、不採算品再算定および最低薬価の充実に向けた検討も行う必要があると考えます。
私からは以上でございます。
それでは中山会長、新薬に関する陳述をお願いいたします。

〇日本製薬工業協会・中山讓治会長(第一三共代表取締役会長)
続きまして、日本製薬工業協会の会長を務めております中山でございます。
スライド10をご覧ください。
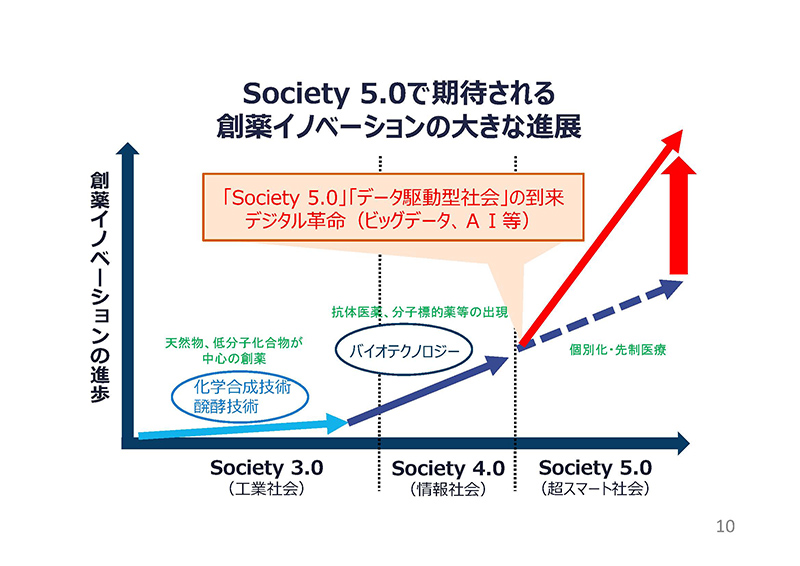
これまで製薬産業は、さまざまな最先端の科学技術を取り込むことで革新的医薬品を世に送り出し、人々の健康や医療の質の向上に貢献してまいりました。
モダリティも、低分子からバイオ医薬品に変化し、個別化医療や再生医療など新しいものが次々と生み出されております。創薬イノベーションは着実に進歩してきたと考えております。
また、これからはビッグデータ、AI、ロボットなど、近年急速に進展しているイノベーションを、あらゆる産業や社会生活に取り入れ、さまざまな社会課題を解決する「Society 5.0」の時代を迎えます。
このデジタル革新は、遺伝子情報の活用による創薬の加速や、治療効果の予測などを可能とし、ヘルスケア分やのイノベーションを大きく進めていくものだと考えております。
スライド11をお願いします。
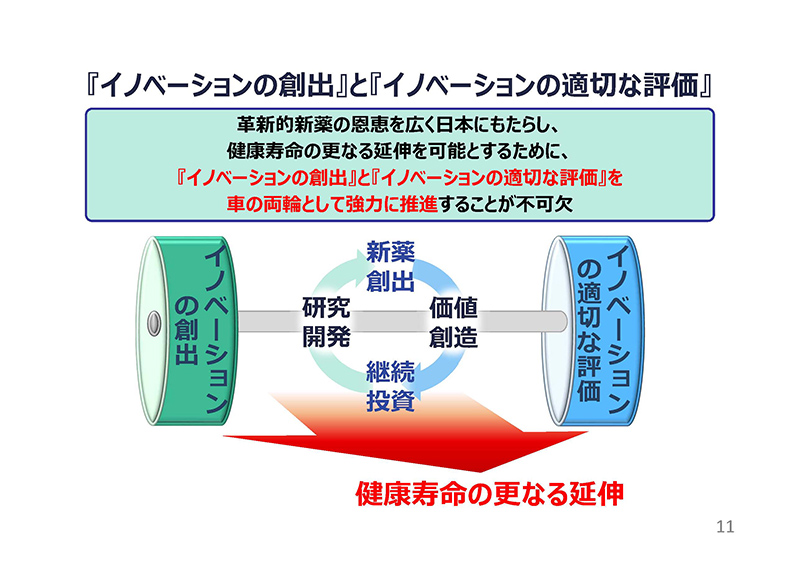
革新的新薬の恩恵を広く日本にもたらし、健康寿命のさらなる延伸を可能とするためには、一方で「イノベーションの創出」の支援と同時に、「イノベーションの適切な評価」の両方を強力に推進することがあってはじめて進むものだというふうに考えております。
スライド12をご覧ください。

イノベーションの適切な評価を行う上で、薬価制度は極めて重要な役割を果たしており、中でも新薬創出等加算は革新的新薬を継続的に創出していくために必要不可欠な仕組みであると考えております。
しかしながら、薬価制度の抜本改革によって対象品目が絞り込まれるとともに、多くの企業の対象品目の薬価が維持されない仕組みとなりました。
次期薬価制度改革においては、真に革新的新薬の創出を促進する仕組みへと改善すべく、新薬創出等加算の品目要件を拡充するとともに、企業要件の見直しを行うべきというふうに考えております。
スライド13をご覧ください。
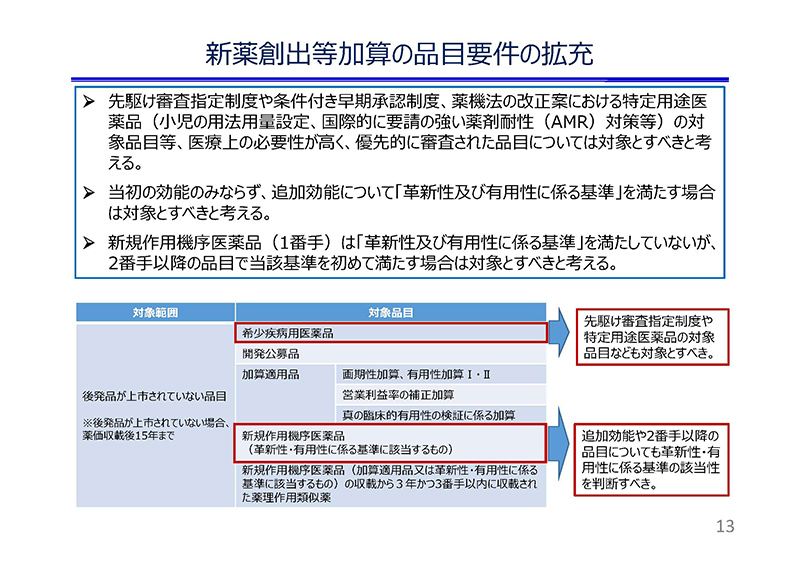
品目要件につきましては1点目として、承認審査において優先的に審査される品目、すなわち「先駆け審査指定制度」や「条件付き早期承認制度」、さらに薬機法の改正案における「特定用途医薬品」等を対象とすべきと考えております。
2点目として、当初の効能のみならず追加効能について、「革新性及び有用性に係る基準」を満たす場合は対象とすべきと考えております。
3点目。現行ルールでは、1番手(新規作用機序医薬品)が「革新性及び有用性に係る基準」を満たさない場合には、2番手以降の品目について当該基準への該当性の判断自体がなされないことになるため、
2番手以降の品目であっても、同一の薬理作用を有する医薬品の中で、当該基準を初めて満たす場合には対象とすべきと考えております。
これらの見直しにより、医療上、開発が強く求められている医薬品や有用な効能追加、既存の治療薬にはない特殊性を備えた医薬品等のさらなる開発の促進につながるものと考えております。
スライド14でございます。
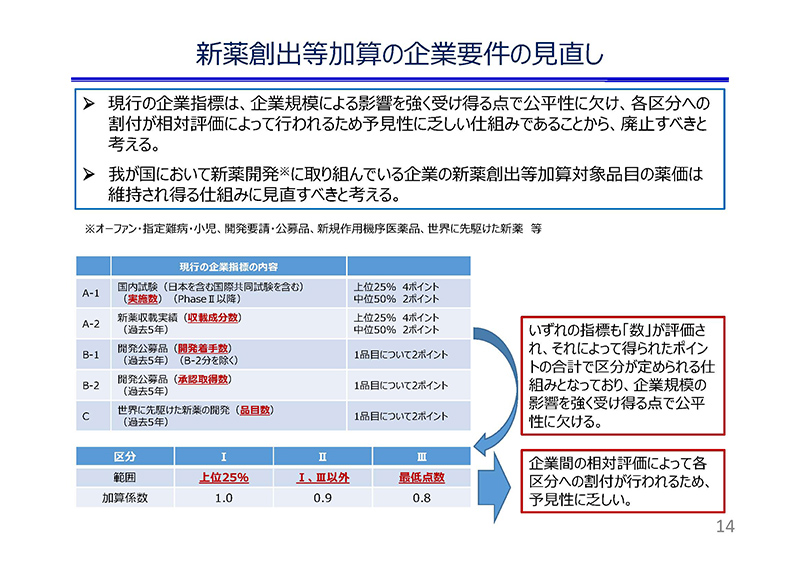
薬価制度改革の抜本改革により新設された企業指標については、
企業規模などの影響を大きく受けるために公平性に欠けること、
また、各区分が相対評価で決まることから予見性に乏しいこと、
などの課題があると認識しております。
従いまして、現行の企業指標は撤廃した上で、日本国内で新薬創出に取り組んでいる企業の品目の薬価が維持される仕組みに見直すべきと考えます。
次に、スライド15です。
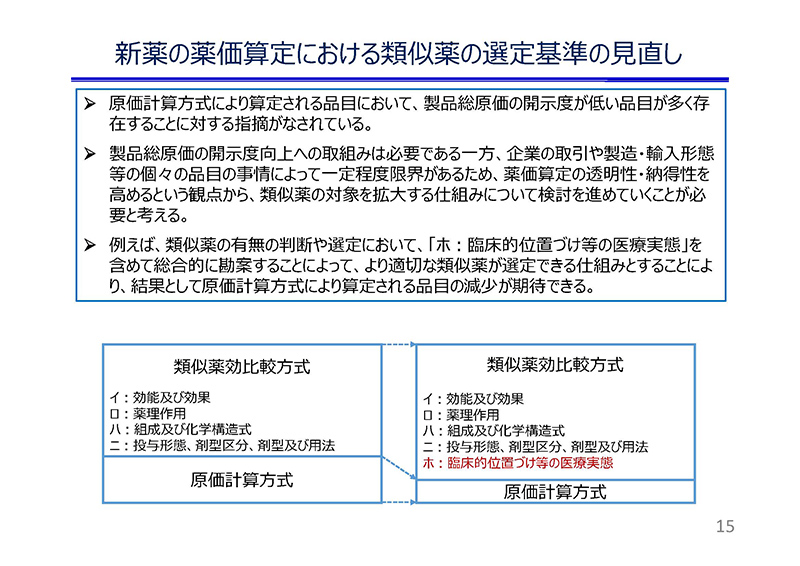
新薬の薬価算定における類似薬の選定基準についてでございます。
現行の薬価制度においては「類似薬効比較方式」を基本とし、適切な新薬が存在しない場合に「原価計算方式」を用いて新薬の薬価が算定されています。
このうち、特に原価計算方式については「製品総原価の開示度」について指摘がなされており、薬価算定の透明性、納得性を高めるという観点から、類似薬の対象を拡大する仕組みの検討が必要であると考えております。
例えば、特定の遺伝子変異の共通性があれば、がん種を問わず投与可能な薬剤がすでに登場してきており、このような新薬については、同じがん種内で類似薬を選定するというルールに縛られることなく、がん種を越えて適切な類似薬を総合的に選定できるルールが必要と考えます。
こうした課題を解決するためには、既存の選定基準にとらわれず、対策疾患の特性、臨床的位置付けなどの「医療実態の類似性」についても総合的に勘案できる仕組みを検討する必要があると考えております。
スライド16をご覧ください。

新薬の薬価収載時の有用性加算の見直しについてでございます。
具体的には、現行の有用性系加算に、医療従事者における負担軽減・リスク軽減の観点が含まれるキット加算を統合し、
製剤工夫等の有無に関わらず、患者・医療従事者双方における治療上の負担軽減、治療の質向上に資する医療的価値を評価できるよう、要件の見直しについて検討が必要を行う必要があると考えております。
例えば、投与の簡便性が向上したり、投与の煩雑な管理が不要になるなど、患者さんの負担軽減を通じてアドヒアランスを高めた医薬品や、
モニタリングが不要であったり、感染の危険を軽減するなど、医療従事者の煩雑な業務を減らすことによって治療におけるリスクを軽減し、患者さんに安全な治療を提供することを可能とする医薬品などがそれに該当すると考えております。
これらは、現行ルールにおきましても評価され得るものでございますけれども、
有用性加算において1つの指標として整理・統合することによって、こうした新薬の開発のさらなる促進につながるものと考えております。
続いてスライド17をご覧ください。
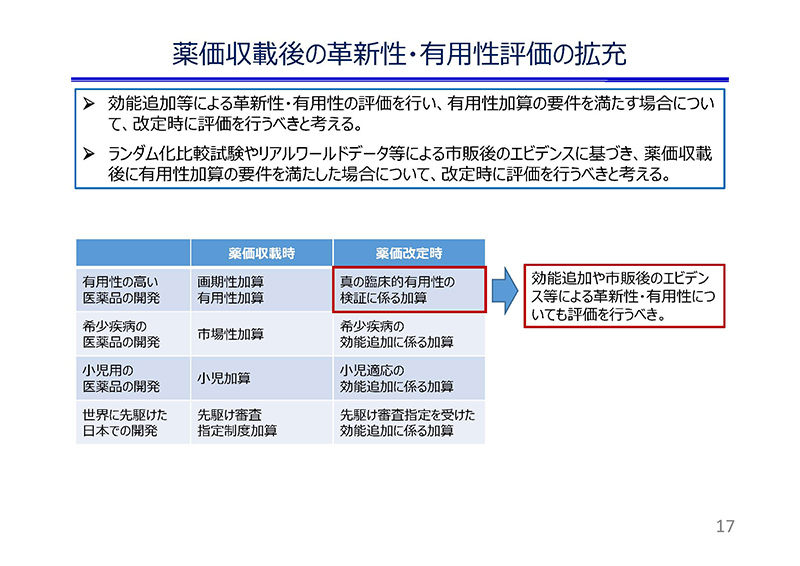
薬価収載後の革新性・有用性評価の拡充についてでございます。
医薬品の中には、薬価収載後に追加した効能によって、新たな革新性や有用性が示される場合がありますが、現行の薬価制度では、それを適切に評価する仕組みとはなっておりません。
一方、既に薬事承認を受けている効能・効果について、市販後に集積された調査成績により、トゥルーエンドポイントが検証された場合の評価として、薬価改定時の真の臨床的有用性の検証に関わる加算がありますが、その適用は極めて限定的でございます。
患者の治療選択肢を増やし、医療の質の向上に貢献し得る効能追加に関わる開発がより促進されるよう、効能追加等によって薬価収載時の有用性系加算の要件を満たす場合には、薬価改定時にその評価を行うべきと考えております。
また、薬価収載時に十分なエビデンスが得られていなかった品目でも、市販後のエビデンス集積によって、新規収載時の有用性系加算の要件を満たすような場合については、薬価改定時に評価を行うべきではないかと考えております。
私からは以上でございます。澤井会長、よろしくお願いします。
〇日本ジェネリック製薬協会・澤井光郎会長(沢井製薬代表取締役社長)
日本ジェネリック製薬協会の澤井でございます。
スライド18をご覧ください。
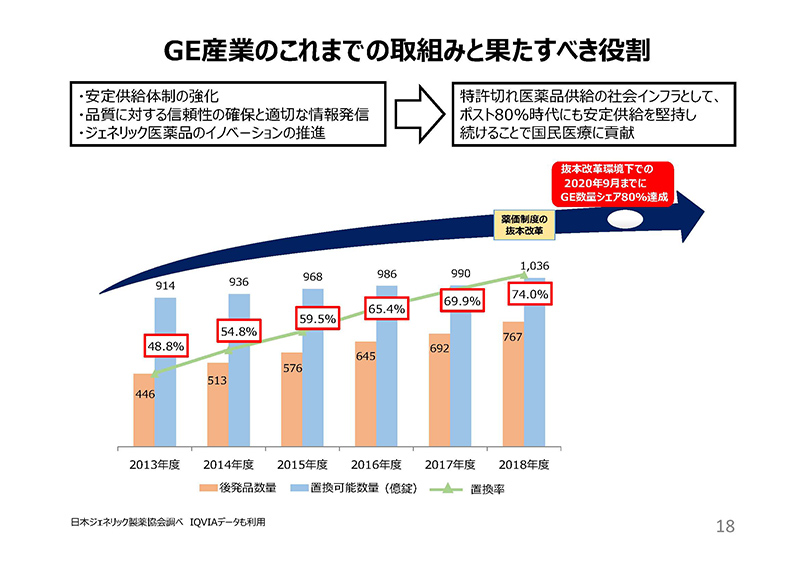
後発品企業は、薬価制度の抜本改革に示された国民負担の軽減と医療の質の向上に貢献するため、数量シェア80%目標の早期達成に向け、これまでさまざまな努力を行ってまいりました。
安定供給体制の強化に向け、業界を挙げて相当の設備投資を行い、原薬のダブルソース化などの取組を行っております。
 品質に対する信頼性の確保と適切な情報発信として、海外原薬製造工場への査察の強化や、原薬製造国の公開、またインタビュー候補に共同開発に関する情報の記載等を行っております。
品質に対する信頼性の確保と適切な情報発信として、海外原薬製造工場への査察の強化や、原薬製造国の公開、またインタビュー候補に共同開発に関する情報の記載等を行っております。
ジェネリック医薬品のイノベーションの推進として、患者のQOLの向上などに貢献する製剤工夫や、包装仕様の改善などを行ってまいりました。
当協会の調べによる2018年度の後発品の数量シェアは74.0%です。
現在、次世代産業ビジョンを策定中ですが、その中においては揺るぎない安定供給体制と、高度な品質管理体制を土台に、品質・安全性情報の提供を行っていくことで、国民医療を守る社会保障制度の持続性に貢献することを目指しております。
2020年度の薬価制度改革は、後発品への置換えの動きを停滞させるものではなく、ポスト80%時代にも特許切れ医薬品供給の社会インフラとして、安定供給を堅持し続けることが可能な制度であるべきと考えております。
スライド19は、後発品の薬価の在り方についてです。

初収載の薬価についてですが、後発品の開発は当該製品の医療ニーズを踏まえ、長期的な安定供給対応を含めた設備投資費用も考慮し、決定しております。
抜本改革の影響が不透明な中で、初収載の薬価がさらに引き下げられることとなれば、後発品が収載される機会が失われると考えます。
後発品企業は、多くの低薬価な医薬品を供給していく使命を担っております。
そのコストを確保する上では、新規収載の薬価が極めて重要です。数量シェア80%となる後発品の安定供給の必要性を踏まえると、初収載薬価は見直すべきではないと考えております。
次に、既収載品の価格帯についてです。
現在の3価格帯および、さらなる価格帯の集約は、市場実勢価格の安いものの薬価が引き上げられたり、逆に、高いものの薬価がその影響で著しく引き下げられるなど、市場実勢価格と改定薬価に乖離が生じております。
抜本改革により導入された、収載から12年経過した後発品の1価格帯への集約化では、多くの品目においてこのような状況が起こると予測されます。
現在の後発品の薬価は、長期的な安定供給を考えず低価格で販売されているような品目も、ひとまとめにされます。
特許切れ医薬品供給の社会インフラとして安定供給を堅持し続け、医療安全のための工夫や患者のQOLの改善などに貢献している医薬品を長期的に供給するためにも、
同一価格帯の中で、改定後の薬価が改定前の薬価を超える品目は別の価格とするなど、市場での評価が適切に反映される制度とすべきと考えます。
以上でございます。
〇日本製薬団体連合会・多田正世会長代理(大日本住友製薬代表取締役会長)
私どもの意見陳述は以上となりますが、
薬価算定ルール見直し等に関する意見の詳細と
製剤の特殊性を踏まえた個別要望事項
につきましては、別添の意見書に取りまとめておりますので、
今後の議論においてご勘案いただければ幸いでございます。
どうも、ご静聴ありがとうございました。

〇米国研究製薬工業協会(PhRMA)在日執行委員会・原田明久副委員長(ファイザー株式会社代表取締役社長)
おはようございます。
PhRMA在日執行委員会で副委員長を務めております原田です。
本日は、次期改定について意見を述べる機会を頂き、誠にありがとうございます。
早速ですが、スライド2をご覧ください。

はじめに、薬価制度抜本改革前後の業界の現状について説明させてください。
 まず、市場の状況ですが、日本の医薬品市場の過去5年間の年平均成長率は1%でありまして、世界の他の地域と比べると、その伸びは低く抑えられております。
まず、市場の状況ですが、日本の医薬品市場の過去5年間の年平均成長率は1%でありまして、世界の他の地域と比べると、その伸びは低く抑えられております。
日本は非常に低い成長率で推移してきたわけですが、PhRMA会員企業はこの間も日本への投資を拡大し、ドラッグラグ問題の解消に積極的に取り組んでまいりました。
これを可能にしたひとつの要因は新薬創出等加算であります。
新薬創出等加算によって特許期間中の薬価が維持され、予見性が高まり、次の新薬開発への再投資にも早期に着手できるという好循環が生まれたためであります。
スライド3をご覧ください。

このような状況の中、2018年度の薬価制度改革におきまして新薬創出等加算の抜本的な見直しが行われました。
また、年4回の再算定や、費用対効果評価による価格調整の導入など、特許期間中の新薬の薬価引下げを加速させる方向の見直しも同時に行われました。
このような、新薬を含む薬価引下げに偏った改革は、研究開発型PhRMA会員企業におきましては、予見性が著しく損なわれ、これまでの好循環を止めることにつながると強く危惧しております。
実際、抜本改革が企業に与える影響を尋ねた調査によりますと、87%の企業が「影響がある」と回答しており、日本の開発優先順位が低下することを危惧する回答が多く寄せられているところであります。
「国民皆保険の持続性」と「イノベーション推進」を本当の意味で両立させ、日本の患者さんに革新的な新薬を今後も迅速に届けることが可能となるよう、次期改定にて薬価制度の改善に向けた検討をお願いしたいと思います。
スライド4をご覧ください。

こちらは、次期改定に向けて検討いただきたい事項のまとめでございます。次のスライド以降で、1つずつ説明させてください。
スライド5をご覧ください。

まず、新薬創出等加算の品目要件についてですが、品目要件の拡充が必要と考えます。
具体的には、拡充を検討いただきたい要件、4点あります。
1点目は、優先審査品目であります。
日本におきましては、幸い関係機関の努力により、承認審査においては、革新的医薬品を世界に遅れることなく早期に承認する体制が整っています。
審査の中で、革新性の高い医薬品とされたものは薬事承認後も、その革新性を評価した新薬創出等加算の対象としていただきたい。
具体的には、先駆け審査指定制度や条件付き早期承認制度、薬機法改正案における特定用途医薬品の対象品目など、医療上の必要性が高く、優先的に審査された品目は対象にすべきと考えます。
2点目は、革新性・有用性の高い効能を追加した品目についてですが、現行の品目要件は「収載時」の評価に偏っており、効能追加などの「収載後」の有用性についても評価すべきと考えます。
3点目は、新薬創出等加算品目を比較薬として算定された新規作用機序医薬品です。
現行の品目要件では、比較薬に対して有用性加算などの補正加算が適用されることが要件の1つになっているわけでありますが、
その有用性の程度が新薬創出等加算品目と同程度と判断され、新薬創出等加算品目が比較薬となった新規作用機序医薬品については、補正加算がなくとも新薬創出等加算の対象にすべきと考えます。
4点目ですが、現行の3番手、3年以内ルールのうち「3年以内要件」の見直しを検討いただきたいと考えております。
同じ薬理作用であっても治療の選択肢が複数存在することは、患者の状態に応じた薬剤選択を可能とする点で医師、患者さん双方にとって重要であると考えます。
これは参考ですが、2000年以降に米国で承認された新規作用機序医薬品のうち、3年以内に2番手が承認された品目の割合は2割以下にとどまっておりまして、
2番手以降の開発が停滞しているという趣旨の分析を最近、FDAが公表しております。
2番手以降の開発を停滞させない観点からも「3年以内要件」について、ご検討をお願いしたいと考えます。
スライド6をご覧ください。
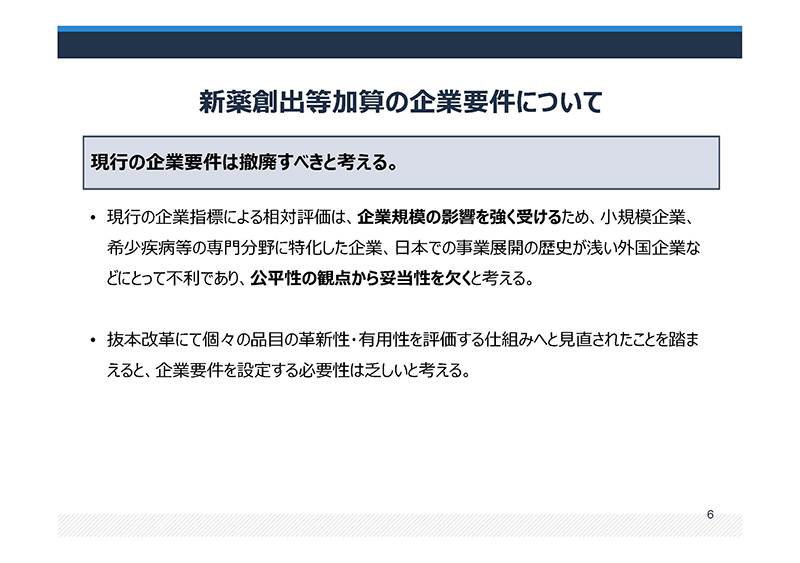
新薬創出等加算の企業要件についてですが、現行の企業要件は撤廃すべきと考えます。
現行の企業指標による相対評価は、企業規模の影響を強く受けるため、小規模企業にとって不利であり、公平性の観点から妥当性を欠くと考えます。
スライド7をご覧ください。

新薬創出等加算対象品目を比較薬とする場合の薬価算定についてです。
新規収載品が新薬創出等加算の対象外であっても、類似薬効比較方式(Ⅰ)で算定される場合には、比較薬の累積加算部分を控除せずに算定されるべきと考えます。
その理由ですが、比較薬と同等の価値を有する新薬の収載時薬価は原則として同じであるべきであり、
比較薬と新薬の間で大きな薬価の差が生じると、市場競争や薬剤選択に対して影響を及ぼす恐れがあると考えます。
スライド8をご覧ください。

改定時の加算についてです。
薬価収載後の革新性を評価する観点から、改定時加算の拡充を検討いただきたいという要望であります。
まず1点目は、革新性の高い効能追加を評価する改定時加算についてです。
現在、効能追加の評価は、小児効能または希少疾病効能等の追加があった場合に限られていますが、
これら以外の効能追加についても、収載時の加算要件に該当するような高い有用性が認められる場合には、改定時加算で評価することを検討いただきたいという要望であります。
2点目は、真の臨床的有用性の対象範囲の拡大であります。
当該加算は現在、有用性の検証が収載後に真のエンドポイントで行われた場合などに非常に限定的に適用されていますが、
収載時には明らかでなかった有用性が、収載後にランダム化比較試験やリアルワールドデータなどで示された場合なども加算対象とすることを検討いただきたいという要望であります。
スライド9をご覧ください。

最後に、再算定についてです。
財政影響の大きい革新的新薬への対応として、再算定ルールの強化が過去数年の間に行われてきました。
さらなる再算定ルールの強化には反対いたします。
また、現行ルールの下では、効能追加が再算定を引き起こす要因となり得ることから、複数の効能に応用可能な革新的新薬では特に、再算定が効能追加への投資判断に影響を及ぼすことになる可能性があります。
本来、効能追加は患者に新たな治療選択肢を提供し、医療の質の向上に資するものであることから、その開発は促進されるべきものと考えます。
再算定が要因となって、効能追加、特に患者数の少ない効能追加への投資判断が影響されることのないよう、開発インセンティブを確保するための処置が必要と考えます。
前のスライドで述べました「効能追加を評価する改定時加算」は、再算定の影響緩和策としてインセンティブになり得るものですので、新設を前向きに検討いただきますよう、よろしくお願いいたします。
PhRMAからは以上です。ありがとうございました。

〇欧州製薬団体連合会のハイケ・プリンツ副会長(バイエル薬品株式会社代表取締役社長)
おはようございます。
[以下、通訳者]

欧州製薬団体連合会副会長のハイケ・プリンツと申します。意見陳述の機会を頂き、ありがとうございます。
スライド2をご覧ください。
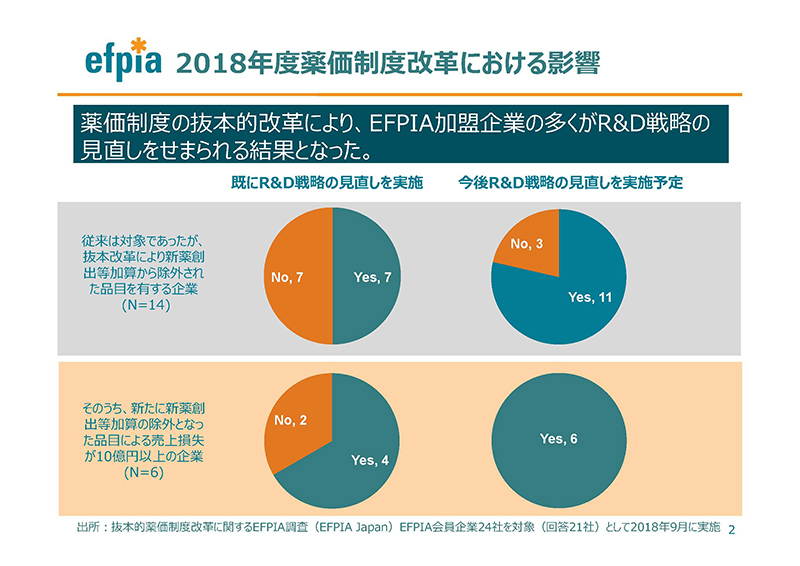
革新性と市場の予見性が適正に評価されることは、われわれ製薬業界が継続してイノベーションを研究開発を通じ、お届けするために必要なことです。
新薬創出等加算は、これが特許保護期間中において新薬の価格の予見性を改善するという意味で、2010年に導入されたあと、製薬業界にとって重要なファクターの1つとなりました。
2010年以降、ドラッグ・ラグの重要な原因の1つと考えられていた申請のラグ、遅れについても著しく改善をしました。
そして国際共同試験を含めて、日本で行われる臨床試験の数は著しく増えています。
また、ほかにも改善された点があります。
(医薬品医療機器総合)機構による審査期間の短縮、あるいは先駆け(審査指定制度)等の新しい審査の仕組みの導入、これらによって画期的な新薬が患者さんに、より早く届けられるようになった事実があります。
しかしながら、2018年に行われた薬価制度の抜本的な改革によって、この環境は全くマイナスの方向に転換してしまいました。
このスライド2のグラフは、私たちが会員企業に対して行った調査に基づくものです。
抜本改革により新薬創出等加算が除外された品目を持つ企業は、そのために研究開発の日本における計画を既に変更したり、あるいは近い将来に変更することを計画しています。
薬価制度が劇的に変わることによって、私どもの海外本社が大きくそれに注目をし、また将来の市場の展望に大きな懸念 を抱いたこと、そして、これが日本の適切なイノベーションの評価の現状についても懸念を持っています。
私たちEFPIAは、もちろん国民皆保険の維持、そしてイノベーションの促進という、この2つの適正なバランスを持ち続けることが必要であることはよく認識しておりますが、
また一方で、2018年に行われた薬価制度の改革がバランスを欠いたものであり、イノベーションの促進という点でマイナスの影響を与えていると考えます。
このあとのスライドで、私たちが今後も継続して画期的な医薬品を日本の患者さんに届け続けるためには、薬価制度においてどのような改善が必要であると考えるか、ご説明いたします。
スライド3をご覧ください。
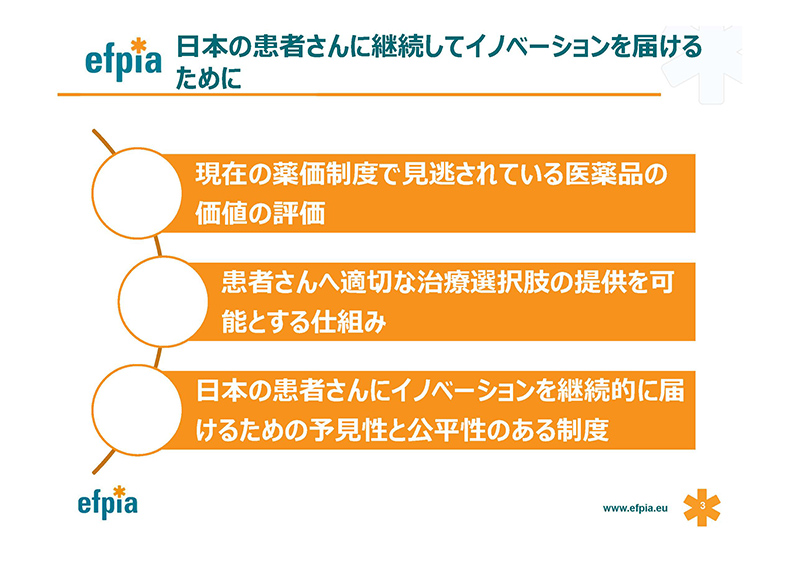
日本のためにイノベーション、画期的な新薬を届け続けるためには何が必要と考えるか、こちらにまとめました。
次のスライドをお願いします。

まず1点目は、この新薬創出等加算についてであります。その品目要件についてであります。
これは現在、新規収載時の、ある一定の価値、そして、その薬理作用1番手の品目の評価に偏っていると考えられます。
私たちがこれに関してお願いすることは、これまでの業界団体が先に説明したとおりであります。
この加算の品目要件につきましても、最初の上市後に追加的な価値が示された品目、そして、それが作用機序の中で1番手でなかったとしても、2番手以降であったとしても、革新性・有用性をある程度示したものには適用されるべきと考えます。
次のスライドをお願いします。
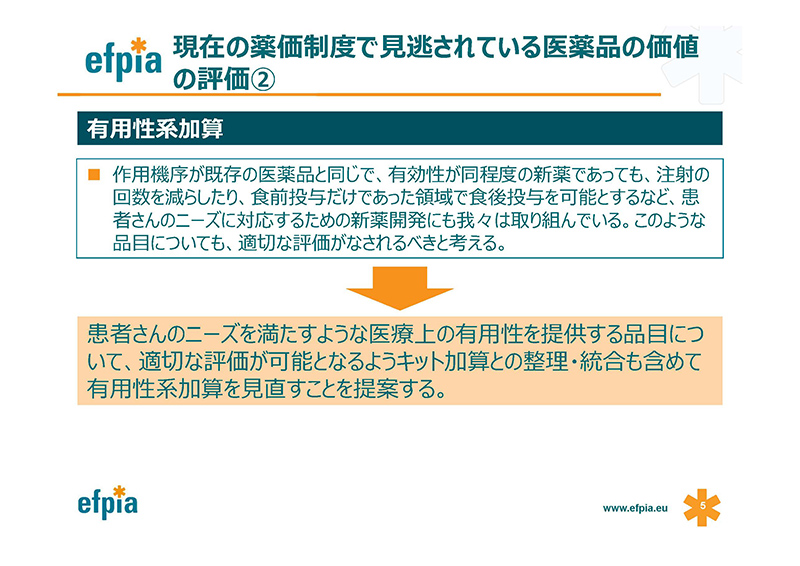
たとえ作用機序が既存の医薬品と同じであったとしても、製薬会社は新薬の開発、研究を続けます。
それは、患者さんのまだ満たされていない医学上のニーズ、あるいは追加的な有用性に応えるためです。
例えば、インスリンのようなホルモン製剤の場合、作用機序は全て同じで、新しい作用機序の品目は存在しません。
例えば、超速効型インスリン製剤の例があります。
患者さんは食事の前に投与されたインスリンの用量に従って、食事で食べる量を調節しなければなりませんでした。
しかしながら、この製剤効果発現までの時間、効果持続時間が短くなることによって食後の注射ができるようになり、実際に食事の量を、食事で摂取した量に合わせて投与することが今、可能になっています。
これは、これまで満たされていなかった医学上のニーズであり、特に食事で摂取する量を予測することが難しいような小児の患者さん、高齢の患者さん、状態の特に悪い患者さんに、特にニーズが高いものです。
この新しい超速効型インスリン製剤のベネフィットによって、このような脆弱な患者さんに実際の食事摂取量に合わせたインスリンの量を投与することができますので、不必要な安全性の懸念を減らすことができます。
こうした品目が適切に評価されるように、現在の有用性加算についてはぜひ見直しをしていただきたい。キット加算との整理・統合も含めて見直しをお願いいたします。
スライド6をご覧ください。

2点目ですけれども、同じ作用機序であっても、ほかの品目の特性、例えば有害事象などの安全性プロファイル、薬物動態のプロファイル、これには半減期ですとか、消失速度、代謝経路などが含まれますが、これらは各薬剤で異なっています。
医師が各患者さんの状態に合わせて適正な薬剤を選択することができるためには、一定の数の異なる薬剤のオプションが存在することが必要だと考えます。
そのオプションの中から選択をする際に、もし、その薬価に大きな違いがあるとすれば、そのときに実際の薬剤の特性ではなく、薬価に基づいて、薬価の違いがその選択に影響を与えてしまう可能性があります。
それに加えて、類似の薬理作用を持つ新薬の場合、そして新薬創出等加算累積相当額を控除するという提案がありますが、
そうなりますと、状況によっては、その薬剤の価格が有用性加算を獲得した場合には類似薬よりも高くなる、あるいは類似薬よりも低い価格になる場合もあるということで、同額の価格になり得ない状況をつくってしまいます。
このような仕組みは、米国あるいは、その他主要な欧州国では存在しておりません。
また、米国、欧州、中国などと比べましても、このような変更がされますと、状況としては日本の薬価の予見性が低くなる。従って、その結果、グローバルでの開発の優先度が日本において影響を受けてしまうという可能性もあります。
従いまして、類似薬効比較方式(Ⅰ)を用いて薬価が決定される新薬につきましては、新薬創出等加算累積相当額の控除は行わず、比較薬と同額での保険償還を可能とすべきと考えます。
スライド7をご覧ください。
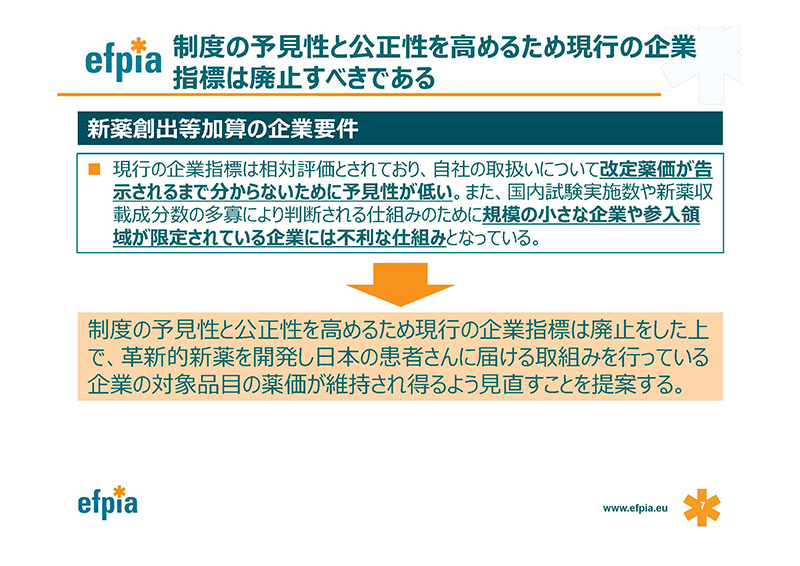
最後に、3点目であります。
この企業要件に関するわれわれの基本的な理解は、先の団体から説明がありました理解と同じであります。
私たちから提案したいことは、この企業要件を見直していただきたい。
そして現在、日本の患者さんへの早期のアクセスができるように画期的な新薬の開発に投資を行っている企業の新薬創出等加算対象品目については、それが維持されるようにしていただきたいとお願いいたします。
ご静聴ありがとうございました。
〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
はい、ありがとうございました。
それでは次に、日本医薬品卸売業連合会より、お願いいたします。
では渡辺会長、お願いいたします。

〇日本医薬品卸売業連合会・渡辺秀一会長(メディパルホールディングス代表取締役社長)
日本医薬品卸売業連合会会長の渡辺でございます。
当連合会を代表して、薬価制度改革に関する意見を述べる機会を賜り、中医協および厚生労働省の皆さまに感謝いたします。
本日は、
Ⅰ 医療保険制度における医薬品卸の役割
Ⅱ 安定的な医薬品流通の確保
Ⅲ 消費税引上げに伴う薬価改定から生ずる課題
Ⅳ 流通改善ガイドラインの遵守状況と今後の課題
の4点について意見を述べさせていただきます。
1ページをご覧ください。

はじめに、「医療保険制度における医薬品卸の役割」についてでございます。
 医薬品卸は、医療保険制度の下、自然災害・パンデミック時を含め、医薬品を安全かつ安定的に供給し、国民医療の向上のため、一定の役割を果たしております。
医薬品卸は、医療保険制度の下、自然災害・パンデミック時を含め、医薬品を安全かつ安定的に供給し、国民医療の向上のため、一定の役割を果たしております。
医薬品卸は、薬事制度や医療保険制度の下で、医薬品の安全確保に努めております。
また、国内24万軒の医療機関、保険薬局との間で、約1万6千品目の医薬品を安定的に供給するとともに、全ての医療機関・保険薬局との間で、早期妥結・単品単価契約を念頭においた価格交渉を行っております。
さらに薬価調査について、任意に医療機関等への納入価格を全て提供し、協力しております。
このような医薬品の安全かつ安定的な流通を通じて、医薬品卸は国民の命や健やかな暮らしを支える医療に貢献することが使命であると考えております。
2ページ目をご覧ください。

次に、「安定的な医薬品流通の確保」についてでございます。
近年の薬価制度改革等によって、医薬品流通を取り巻く環境が大きく変化しており、安定的な医薬品流通の確保に関して、次のような懸念があります。
本年10月には、消費税引上げに伴う薬価改定、来年度は2年に1度の薬価の通常改定が予定されており、そして2021年度には中間年の薬価改定が予定されております。
医薬品卸は、医療保険制度の根幹にかかわる薬価調査の信頼性確保にも資するよう、人的資源を最大限に投入して早期妥結を念頭においた価格交渉を精力的に行っており、そのために多大な労力を費やしております。
また、医薬品卸には、流通改善ガイドラインに則して流通改善を進めつつ、今後も平時はもとより、自然災害やパンデミック時においても医薬品の安定供給を確保することが求められております。
しかしながら、薬価調査や薬価改定に関わる医薬品卸への急激な負担の増大は、これらの卸の役割を大きく損い、ひいては医薬品の安定供給に支障を生じさせかねません。
新薬創出加算品目適用範囲の縮小や、後発品の使用促進策に伴う長期収載品の厳しいルールの適用などにより、医療用医薬品の市場構造は大きく変化しており、市場の成長が過度に抑制されれば、医薬品の配送体制の強化や災害時への備えなどに影響を生じかねません。
なお、一部の後発品や基礎的医薬品については、これ以上薬価が下がることとなれば、医薬品の安定供給に支障を生じかねないのではないかと懸念しております。
薬価制度改革に当たっては、過度に財政を優先することなく、医薬品の安定供給等の卸の取組や役割に配慮していただき、安定的な医薬品流通に支障が生ずることないよう、慎重に検討をお願いいたします。
3ページ目をご覧ください。
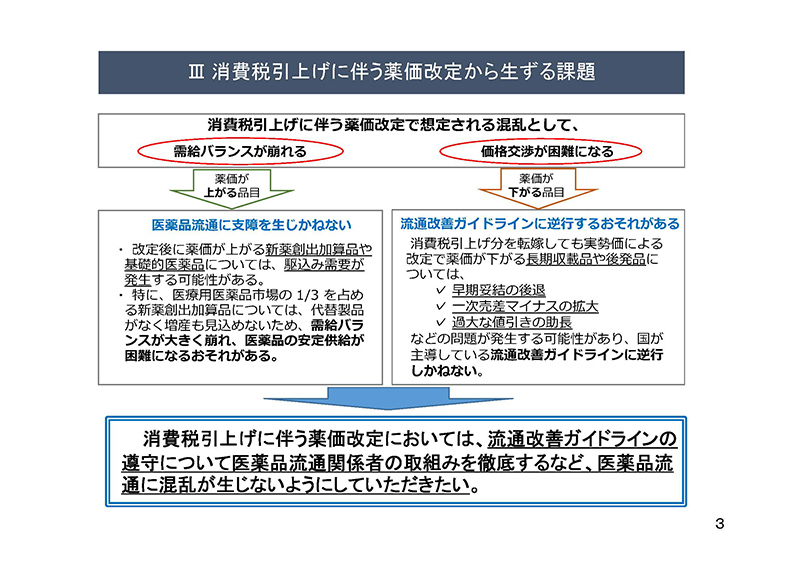
「消費税引上げに伴う薬価改定から生ずる課題」についてでございます。
本年10月には消費税引上げに伴い、近年例のない年度途中の薬価改定が予定されております。
今回の薬価改定では、薬価が上がる品目や下がる品目が混在し、それぞれの品目について需給状況の変動や、取引先との価格交渉が困難になるなど、医薬品流通の混乱が見込まれます。
具体的には、薬価が上がる新薬創出加算品や基礎的医薬品については駆込み需要の発生が考えられます。
特に、新薬創出加算品については、代替製品がなく増産も見込めないため需給バランスが大きく崩れることとなれば、医薬品の安定供給が困難になる恐れがあります。
また、消費税引上げ分を転嫁しても薬価が下がる薬価が下がる長期収載品や後発品については、早期妥結の後退や過大な値引きを助長しかねない。このことに伴って、一次売差マイナスの拡大を招く可能性があり、国が主導する流通改善ガイドラインに逆行
しかねないかと危惧をしております。
このような課題に対応するため、消費税引上げに伴う薬価改定においては、流通改善ガイドラインの遵守について医薬品流通関係者の取組を徹底するなど、医薬品流通に混乱が生じないようにしていただきたいと考えております。
4ページをご覧ください。

最後に、「流通改善ガイドラインの遵守状況と今後の課題」についてでございます。
2018年度の薬価制度の抜本改革の骨子では、2021年度を初年度とする中間年の薬価改定に向けて、安定的な医薬品流通が確保されるよう、単品単価契約、早期妥結等を積極的に推進するとされております。
流通関係者は、流通改善ガイドラインに即した取組を行い、流通改善に一定の進展が見られましたが、今後、改善すべき点があります。
早期妥結については、未妥結減算制度と相まって、9月までの妥結率が大幅に向上し、遡及値引きがなくなるなど、進展が見られました。
しかしながら、部分妥結や9月までの妥結価格が10月以降の再交渉で変動する取引など、一部に未妥結減算や流通改善ガイドラインの趣旨に反する取引が行われており、改善が必要であると考えております。
単品単価契約率については、流通改善ガイドラインに即した流通関係者の取組などにより、単品単価契約率の大幅な上昇が見られました。
引き続き、価格交渉の段階から医薬品の価値を踏まえた交渉を推進していくため、薬価の本体価格の値引き率による交渉の徹底に向けて、さらに取り組んでいく必要があると考えております。
今後、未妥結減算制度や流通改善ガイドラインの趣旨が徹底されるようにお願いいたします。
以上、日本医薬品卸売業連合会としての意見を述べさせていただきました。何卒、よろしくお願い申し上げます。
〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
はい、ありがとうございました。
それでは次に、再生医療イノベーションフォーラム(FIRM)および日本バイオテク協議会より、お願いいたします。
ではまず、畠会長からお願いします。

〇再生医療イノベーションフォーラム・畠賢一郎代表理事会長(ジャパン・ティッシュエンジニアリング代表取締役会長執行役員)
再生医療イノベーションフォーラム、FIRMの会長をしておりますジャパン・ティッシュエンジニアリングの畠でございます。
本日はこのような発言の機会を頂きまして、誠にありがとうございます。
FIRMは、再生医療を対象とした唯一の業界団体でありますので、再生医療等製品の価格算定に関しまして、意見を述べさせていただければと思っております。
まずはじめに、2014年11月に旧薬事法が改正されまして「医薬品医療機器等法」が成立いたしました。医薬品医療機器とは別に、再生医療等製品として分類していただきましたことを深く関係各位に感謝申し上げます。
施行されて約5年が経過いたしますが、別に分類されましたことで、その特性を踏まえた相談制度や審査制度の充実が図られているものと実感をしております。
2ページ目をご覧ください。

まずはじめに、再生医療等製品の特徴について、ご説明させていただきます。
再生医療等製品は、今まで治療不可能であった疾患を治療可能とするなど、今までとは違った治療概念を持つ、そういった治療概念を大きく変える可能性があるものだという認識をしております。
また、単回投与あるいは1回の移植で、その効果が長期にわたり持続する可能性があるものでございます。患者さまにもたらす利益は非常に大きいものと期待されております。
しかしながら、ヒト細胞を利用したものといっても、極めて多種多様でございます。製品ごとに、その製造工程や品質管理は異なってまいります。また、その対象患者さんも限定されるのが現状でございます。
さらに、製品自体のシェルフライフ(使用・有効期限)が極端に短い場合、移植までに時間制限がありましたり、それから製品によりましてはですね、事前に先生方にトレーニングを受けていただく必要があったり、また施設が限定されることもございます。
さらにですね、サプライチェーンが非常に複雑でございまして、その中には多くの知財が含まれることもありまして、1企業で賄うことが難しい側面もございます。
3ページをご覧ください。

さて、再生医療等製品のコストについてでございます。
再生医療等製品は、ロットを構成しないことが多かったりですね、また1ロットが少ないケースが多く、技術的にスケールメリットを出しにくいのが現状でございます。
また、施設・設備の転用が極めて難しゅうございます。
その設備コストや多種多様な検査コスト、特殊な保管や輸送コストが必要になってまいります。
さらに、遺伝子組み換え製品でございますが、ウイルスの拡散防止対策も必要となっております。
また、製造工程でございますが、高度な細胞培養技術を持つ人材の育成・確保が必要になっております。
さらに、細胞培養などには大変多くの知財が存在しております。そのライセンス料が必要となる場合もあります。
4ページをご覧ください。
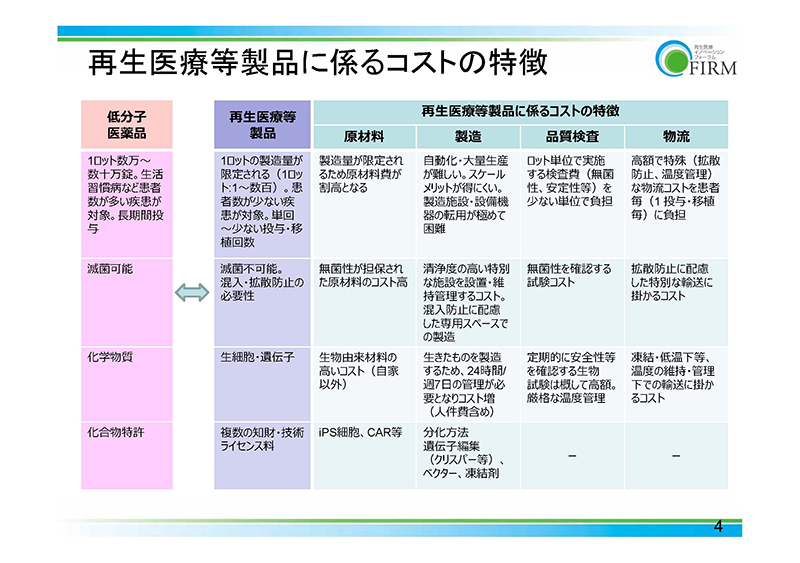
この表は、先ほどお話ししました内容を含めまして、コストの特徴を低分子医薬品と比較して一覧にしたものでございます。それぞれの項目につきまして、再生医療等製品と対比いたしました。
それぞれについて、原材料、製造、品質検査、物流、そういったものに関するコストの特徴を記しております。ご参考になればというふうに思います。
それでは、5ページをご覧ください。

ここまで、お話しさせていただきましたように、再生医療等製品は厳重な製造工程管理管理、それから品質管理が必要でございます。
生産の効率化が現状のところ非常に厳しく、医薬品や医療機器とはコスト構造が大きく異なってまいります。
従いまして、現行の算定方法では再生医療等製品の特殊性を適切に反映していただけないのでは、というふうに考えております。
新たな治療方法によりまして、患者さまへの貢献が期待される再生医療等製品でございますが、
薬機法(医薬品医療機器等法)と同様にですね、医薬品・医療機器とは別のカテゴリーとして分けていただき、その革新性・画期性が十分に反映できる価格算定方式を検討していただければと切に願う次第でございます。
どうぞよろしくお願いいたします。
最後でございます。6ページ目でございます。
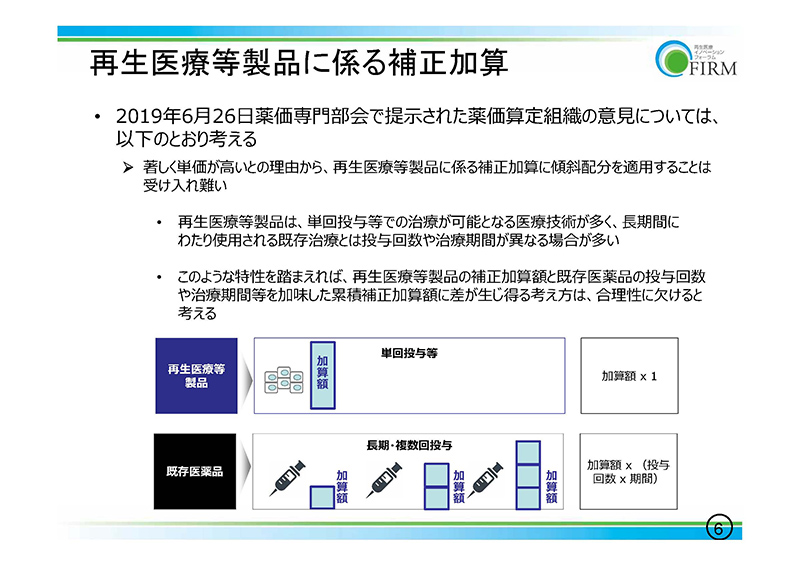
先日の薬価専門部会で提示されました「著しく単価の高い再生医療等製品は、補正加算率を傾斜配分してはどうか」というご意見を賜っておりますが、
詳細は時間の関係上、ご説明できませんが、当業界団体としましては、ここに記載させていただきました理由からですね、受けることが困難だろうというふうに考えておりますので、ご配慮いただければというふうに思っております。
以上、ご説明させていただきました。ご静聴ありがとうございました。よろしくお願いいたします。
〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
では山田会長、よろしくお願いいたします。

〇日本バイオテク協議会・山田英会長(アンジェス株式会社代表取締役社長)
 一般社団法人・日本バイオテク協議会会長の山田でございます。
一般社団法人・日本バイオテク協議会会長の山田でございます。
本日は、意見陳述の機会を頂き、誠にありがとうございます。
現在、会員企業は45社となっており、イノベーティブな難病薬や希少疾病薬の開発を通じて、国民医療水準の向上に貢献しております。
それでは理事の塩村より、私たちの主張、提案を説明させていただきます。
〇日本バイオテク協議会・塩村仁理事(ノーベルファーマ株式会社代表取締役社長)
理事の塩村でございます。
2ページをご覧ください。

バイオテク協議会の会員企業の開発医薬品は、革新性が高い難病・希少疾病用薬が多く、現行新薬算定ルールで想定外の事例が散見されております。
 3つ挙げます。
3つ挙げます。
1番。
医薬品条件付早期承認制度などの積極的活用は、新薬アクセスにとって非常に重要であります。
しかし、検証的臨床試験結果のない新薬について、その有用性が薬価算定において評価されず、この素晴らしい制度の活用をためらうケースが出ております。
2番。
既存薬を全く異なる用途で開発する。
例えば昔、アスピリンというのが消炎鎮痛剤として知られておりましたが、現在では、血栓抑制の薬剤というポジショニングになっておりますけれども、
こういうものをドラッグリポジショニングと言いまして、これはアカデミア、創薬ベンチャーでよく使われる新薬の方法であります。
後ほど述べますけれども、現行特例算定ルールでは極端な低薬価となり、必要な開発経費、市販後安全対策、安定供給等にかかわる最低限の経費を確保できないため、開発着手されなくなっております。
3番目。
世界初の革新的医薬品を日本から上市すると、何かと不利な点がございます。創薬ベンチャーの多くが海外展開を視野に入れておりまして、本邦の薬価が低薬価となりますと海外も引きずられるということになります。
日本を後回しにして海外先行開発という、おかしな事態を招きかねないというふうに心配しております。
3ページをご覧ください。

難病・希少疾病用薬の薬価算定に対することとして、創薬ベンチャーはこういうものの開発が多いんですけれども、患者数が限定的であり、高薬価でないと採算が取れません。
患者1人当たりの薬価が高額で、保険財政に大きな影響があるように誤解されますが、実際には市場規模が小さく、医療費に与える影響は大きくありません。
先ほど申しましたように、日本での薬価が諸外国より低ければ、日本発医薬品でも海外先行開発を誘引すると。
とりわけ、ウルトラオーファン薬につきましては、高額算定が予見されないと開発意欲を削ぐということになります。
4ページをご覧ください。

医薬品の条件付早期承認制度などについて、条件付加算および、収載後、評価を見直して、場合によっては引き下げる、あるいは引き上げるということもやっていただければというふうに考えております。
ドラッグリポジショニング新薬の特例算定について改定をお願いしたいと。これは後ほど、6ページ、7ページでご案内をさせていただきます。
ウルトラオーファン加算の新設をお願いいたしたい。これは13ページに詳しく書いておりますので、後ほどご覧ください。
5ページをご覧ください。

ドラッグリポジショニング新薬の特例算定の実情がこのページに書いてあります。
簡単に言いますと、原価計算方式または類似薬効比較方式のうちの価格の低いほうに決めるという算定ルールでございますけれども、
参考の11ページにありますように、

古い薬でございますので、原価も低くなっていて、非常に低い薬価になってしまうということで、もう現在、なかなかこの手法を使って開発するということは、ためらう状態になっております。
6ページをご覧ください。

これについて改定をお願いしたいというのが6ページの一番下に書いてございます。
同一成分の既収載品が極端な低薬価である場合や、難病等患者数が非常に少ない場合には、特例算定を適用せずに通常の算定ルールでお願いしたい。
以上でございます。ご静聴ありがとうございました。

〇中村洋部会長(慶應義塾大大学院経営管理研究科教授)
ありがとうございました。

-1-185x130.jpg)











【速記録】_ページ_01-のコピー.jpg)
【速記録】_ページ_01-のコピー.jpg)
【速記録】_ページ_01-のコピー.jpg)
_2023年8月2日の総会-1-190x190.jpg)

_2023年6月21日の中医協総会-190x190.jpg)








_2022年8月3日の中医協総会-190x190.jpg)















-190x190.jpg)


_20190807_中医協材料ヒアリング-300x300.jpg)





























